Se hai un abbonamento attivo ACCEDI QUI
La sindrome di Miller-Dieker è una rara malattia genetica caratterizzata da lissencefalia di tipo 1 e tratti facciali distintivi. Questa condizione è causata da una delezione nella regione cromosomica 17p13.3, che include il gene LIS1, fondamentale per la migrazione neuronale durante lo sviluppo cerebrale. I pazienti affetti presentano gravi ritardi nello sviluppo psicomotorio, convulsioni e difficoltà alimentari. La prognosi è generalmente sfavorevole, con una sopravvivenza limitata all’infanzia o alla prima infanzia.
Cos’è la sindrome di Miller-Dieker
La sindrome di Miller-Dieker (MDS) è una malattia genetica rara caratterizzata da lissencefalia di tipo 1 e tratti facciali distintivi. La lissencefalia, o “cervello liscio”, è una malformazione cerebrale in cui la corteccia presenta una riduzione o assenza delle normali circonvoluzioni. I pazienti con MDS manifestano gravi ritardi nello sviluppo psicomotorio, convulsioni e difficoltà alimentari. La prognosi è generalmente sfavorevole, con una sopravvivenza limitata all’infanzia o alla prima infanzia.
I tratti facciali caratteristici includono fronte prominente, solchi bitemporali stretti, naso corto con narici all’insù e labbro superiore prominente. Questi segni distintivi possono facilitare la diagnosi clinica precoce. Tuttavia, la conferma diagnostica richiede indagini genetiche specifiche. La MDS è associata a una delezione nella regione cromosomica 17p13.3, che include il gene LIS1, fondamentale per la migrazione neuronale durante lo sviluppo cerebrale.
La prevalenza esatta della MDS è sconosciuta, ma si stima che sia estremamente rara. La trasmissione è generalmente autosomica dominante, sebbene la maggior parte dei casi sia sporadica, derivante da delezioni de novo. La diagnosi prenatale è possibile attraverso tecniche di citogenetica molecolare, come l’ibridazione fluorescente in situ (FISH), che possono rilevare delezioni submicroscopiche nella regione 17p13.3. La consulenza genetica è fondamentale per le famiglie con una storia di anomalie cromosomiche per valutare il rischio di ricorrenza in future gravidanze.
La gestione dei pazienti con MDS è principalmente sintomatica e di supporto. Le convulsioni possono essere trattate con farmaci antiepilettici, sebbene spesso siano resistenti al trattamento. Le difficoltà alimentari possono richiedere l’uso di sondini nasogastrici o la realizzazione di una gastrostomia per garantire un adeguato apporto nutrizionale. Il supporto multidisciplinare, che include neurologi, genetisti, nutrizionisti e terapisti occupazionali, è essenziale per ottimizzare la qualità di vita dei pazienti e delle loro famiglie.
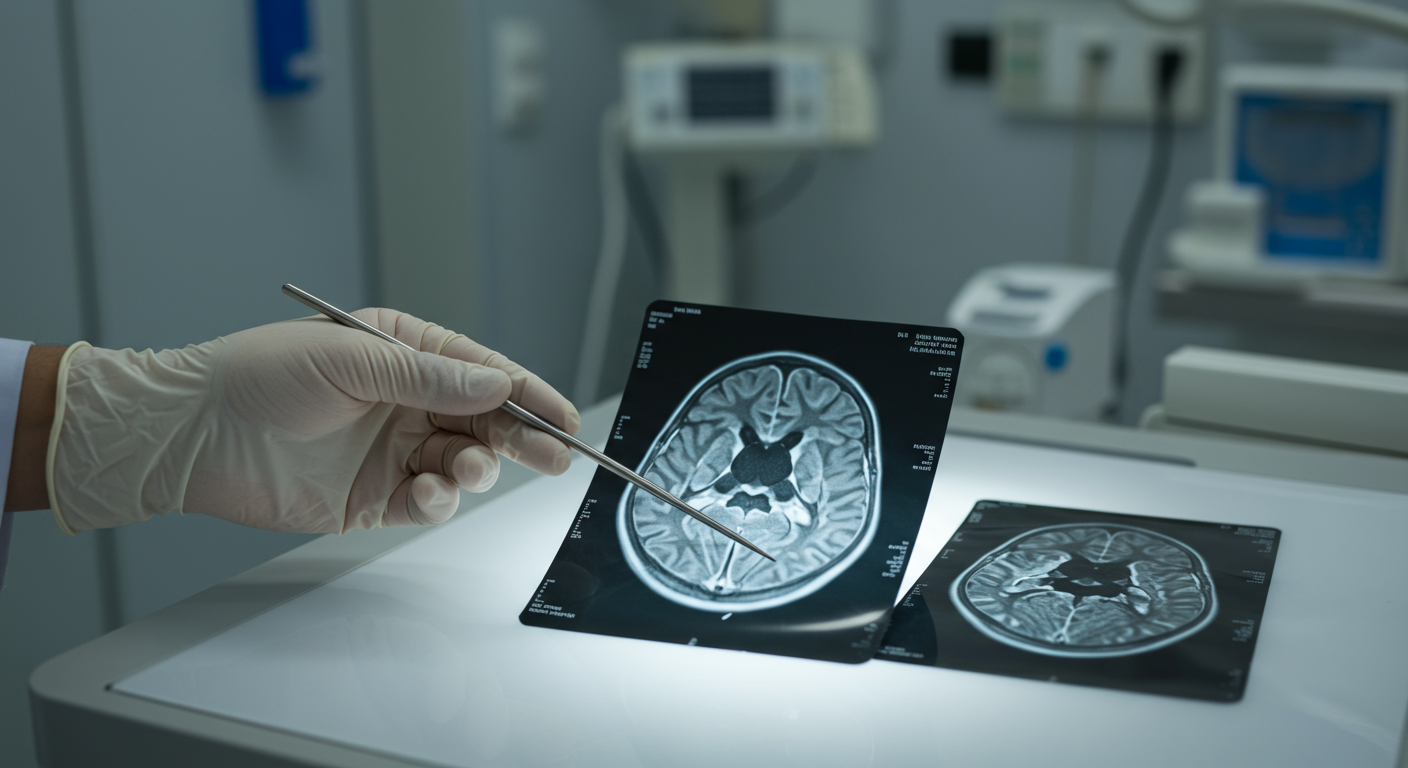
Lisencefalia: cervello liscio senza circonvoluzioni
La lisencefalia è una malformazione cerebrale caratterizzata da una superficie corticale liscia, con assenza o riduzione delle normali circonvoluzioni cerebrali. Questa condizione deriva da un difetto nella migrazione neuronale durante lo sviluppo fetale, che impedisce la formazione dei solchi e delle circonvoluzioni tipiche del cervello maturo. La lisencefalia può presentarsi in vari gradi di severità, da una completa agiria (assenza totale di circonvoluzioni) a una pachigiria (circonvoluzioni insolitamente ampie e spesse). Queste anomalie strutturali compromettono significativamente le funzioni neurologiche, causando gravi ritardi nello sviluppo psicomotorio e disabilità intellettiva.
I sintomi clinici associati alla lisencefalia includono ipotonia muscolare, convulsioni, difficoltà nell’alimentazione e ritardo nella crescita. Le convulsioni, in particolare, sono una caratteristica prominente, manifestandosi nel 90% dei bambini prima dei 6 mesi di età, spesso inizialmente sotto forma di spasmi infantili. La gravità dei sintomi è generalmente correlata all’estensione della malformazione cerebrale. Nei casi più severi, i pazienti possono presentare microcefalia, con una riduzione del volume cranico rispetto alla norma.
La diagnosi di lisencefalia si basa su indagini di neuroimaging, in particolare la risonanza magnetica (RM), che permette di visualizzare l’assenza o la riduzione delle circonvoluzioni cerebrali. L’RM può anche rivelare altre anomalie associate, come l’ipoplasia del corpo calloso o la dilatazione dei ventricoli cerebrali. L’analisi genetica è fondamentale per identificare le mutazioni specifiche associate alla lisencefalia, come quelle nel gene LIS1, che è frequentemente coinvolto nella sindrome di Miller-Dieker. La diagnosi prenatale è possibile attraverso tecniche di citogenetica molecolare, come l’ibridazione fluorescente in situ (FISH), che possono rilevare delezioni submicroscopiche nella regione 17p13.3.
Il trattamento della lisencefalia è principalmente sintomatico e di supporto. Le convulsioni possono essere gestite con farmaci antiepilettici, sebbene spesso siano resistenti al trattamento. Le difficoltà alimentari possono richiedere l’uso di sondini nasogastrici o la realizzazione di una gastrostomia per garantire un adeguato apporto nutrizionale. Il supporto multidisciplinare, che include neurologi, genetisti, nutrizionisti e terapisti occupazionali, è essenziale per ottimizzare la qualità di vita dei pazienti e delle loro famiglie.
Mutazione LIS1 o delezione 17p13.3
La sindrome di Miller-Dieker è principalmente causata da una delezione nella regione cromosomica 17p13.3, che include il gene LIS1. Il gene LIS1 codifica per una proteina essenziale nella migrazione neuronale durante lo sviluppo cerebrale. La perdita di questo gene interrompe il normale processo di migrazione dei neuroni, risultando nella formazione di una corteccia cerebrale liscia, caratteristica della lissencefalia. Oltre a LIS1, la delezione può coinvolgere altri geni nella stessa regione, contribuendo alla variabilità fenotipica osservata nei pazienti con MDS.
Le delezioni nella regione 17p13.3 possono verificarsi de novo o essere ereditarie, soprattutto in presenza di riarrangiamenti cromosomici parentali, come traslocazioni bilanciate. In questi casi, un genitore può essere portatore sano di una traslocazione che, se trasmessa in modo sbilanciato al figlio, può causare la sindrome. La consulenza genetica è fondamentale per le famiglie con una storia di anomalie cromosomiche per valutare il rischio di ricorrenza in future gravidanze. La diagnosi prenatale è possibile attraverso tecniche di citogenetica molecolare, come l’ibridazione fluorescente in situ (FISH), che possono rilevare delezioni submicroscopiche nella regione 17p13.3.
La diagnosi genetica della MDS richiede l’uso di tecniche avanzate, come l’ibridazione fluorescente in situ (FISH) o l’analisi del microarray cromosomico, per identificare delezioni submicroscopiche nella regione 17p13.3. Questi test sono fondamentali per confermare la diagnosi clinica e per fornire informazioni prognostiche. La diagnosi prenatale è possibile attraverso l’analisi del DNA fetale ottenuto tramite amniocentesi o villocentesi, permettendo l’identificazione precoce della delezione cromosomica. La consulenza genetica è fondamentale per le famiglie con una storia di anomalie cromosomiche per valutare il rischio di ricorrenza in future gravidanze.
La comprensione delle basi genetiche della MDS ha implicazioni significative per la gestione clinica e la consulenza genetica. Sebbene attualmente non esista una terapia curativa per la MDS, la diagnosi precoce e una gestione multidisciplinare possono migliorare la qualità di vita dei pazienti.
Ritardo grave e convulsioni
La sindrome di Miller-Dieker è caratterizzata da un marcato ritardo nello sviluppo psicomotorio, che si manifesta fin dai primi mesi di vita. I bambini affetti faticano a raggiungere le principali tappe evolutive, come il controllo del capo, la capacità di sedersi o la produzione di parole. Questo deficit globale è la conseguenza diretta delle anomalie strutturali della corteccia cerebrale e comporta una dipendenza costante dall’assistenza sanitaria e familiare.
Accanto al ritardo psicomotorio, le convulsioni rappresentano una delle complicanze più comuni e invalidanti. Le crisi epilettiche esordiscono spesso nei primi mesi di vita e possono assumere forme diverse, tra cui spasmi infantili, crisi tonico-cloniche generalizzate e crisi focali complesse. La loro frequenza e gravità contribuiscono a peggiorare ulteriormente il quadro clinico e la prognosi generale. (orpha.net)
L’epilessia nella sindrome di Miller-Dieker è spesso farmacoresistente, rendendo complessa la gestione terapeutica. Nonostante l’impiego di farmaci antiepilettici in combinazione, molte crisi possono persistere, richiedendo controlli neurologici regolari e aggiustamenti terapeutici frequenti. La gestione dei sintomi punta a ridurre la frequenza delle crisi, limitare gli effetti collaterali dei farmaci e migliorare il comfort del paziente. (medicoverhospitals.in)
La combinazione di grave ritardo nello sviluppo e convulsioni refrattarie incide profondamente sulla qualità di vita e sulla prognosi. Un approccio multidisciplinare, che coinvolge neurologi, pediatri, fisioterapisti e logopedisti, è essenziale per offrire il massimo supporto possibile. Interventi precoci di riabilitazione, terapie occupazionali e programmi di assistenza personalizzata possono contribuire a migliorare, seppur in modo limitato, le capacità residue e il benessere complessivo del bambino e della sua famiglia.
Diagnosi prenatale e supporto neurologico
La diagnosi prenatale della sindrome di Miller-Dieker (MDS) è fondamentale per una gestione tempestiva e informata della gravidanza. Durante le ecografie prenatali, è possibile identificare anomalie cerebrali indicative di lissencefalia, come l’assenza delle normali circonvoluzioni cerebrali. In presenza di tali sospetti, si raccomanda l’esecuzione di test genetici invasivi, quali l’amniocentesi o il prelievo dei villi coriali, per rilevare delezioni specifiche sul cromosoma 17p13.3. (courses.fetalmedicine.com)
Una volta confermata la diagnosi, è essenziale un approccio multidisciplinare per pianificare il percorso assistenziale del neonato. Il supporto neurologico precoce è cruciale per affrontare le sfide associate alla MDS, tra cui convulsioni, difficoltà alimentari e ritardi nello sviluppo. La gestione delle crisi epilettiche spesso richiede l’uso di farmaci antiepilettici, con monitoraggio regolare per valutare l’efficacia e gli eventuali effetti collaterali. (medicoverhospitals.in)
Le difficoltà alimentari, comuni nei bambini con MDS, possono portare a malnutrizione e complicanze respiratorie. In questi casi, si può ricorrere a soluzioni come l’alimentazione tramite sondino nasogastrico o la gastrostomia, per garantire un adeguato apporto nutrizionale e prevenire l’aspirazione polmonare. (orpha.net)
Il coinvolgimento di un team di specialisti, tra cui neurologi, genetisti, pediatri e terapisti, è fondamentale per fornire un’assistenza completa e personalizzata. Programmi di intervento precoce, terapie fisiche e occupazionali possono contribuire a migliorare le capacità motorie e cognitive del bambino, ottimizzando la qualità della vita.
Per approfondire
Orphanet: Sindrome di Miller-Dieker – Informazioni dettagliate sulla sindrome, inclusi segni clinici, genetica e gestione.
Wikipedia: Sindrome di Miller-Dieker – Panoramica generale sulla sindrome, con dettagli su sintomi, eziologia e prognosi.
FMF Courses: Anomalie fetali – Informazioni sulle anomalie cerebrali fetali, inclusa la lissencefalia associata alla sindrome di Miller-Dieker.
Ospedale Meyer: Neurogenetica – Servizi di diagnosi e gestione per condizioni neurogenetiche, inclusa la lissencefalia.
Malattie Rare Toscana: Lissencefalia isolata o sindromica – Informazioni sulle forme di lissencefalia e percorsi diagnostici disponibili.